Myotonic Dystrophy Type 1 Program

Our DM1 program is designed to address the underlying genetic cause of DM1 with the potential to restore function of an essential protein and its critical downstream products.
Our DM1 program is designed to address the underlying genetic cause of DM1 with the potential to restore function of an essential protein and its critical downstream products.
40,000+ people in the U.S have DM1.1
50,000+ people in Europe have DM1.1
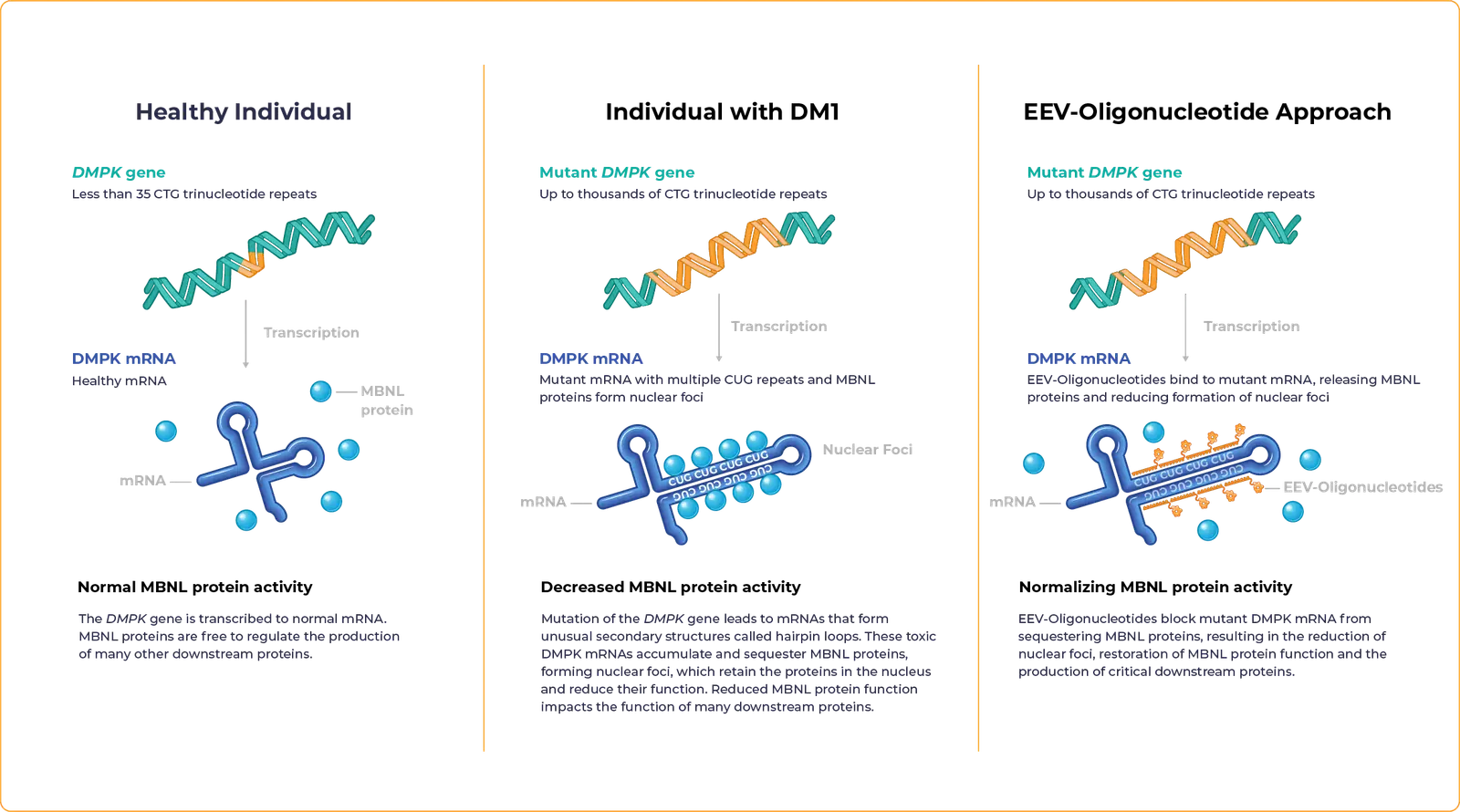
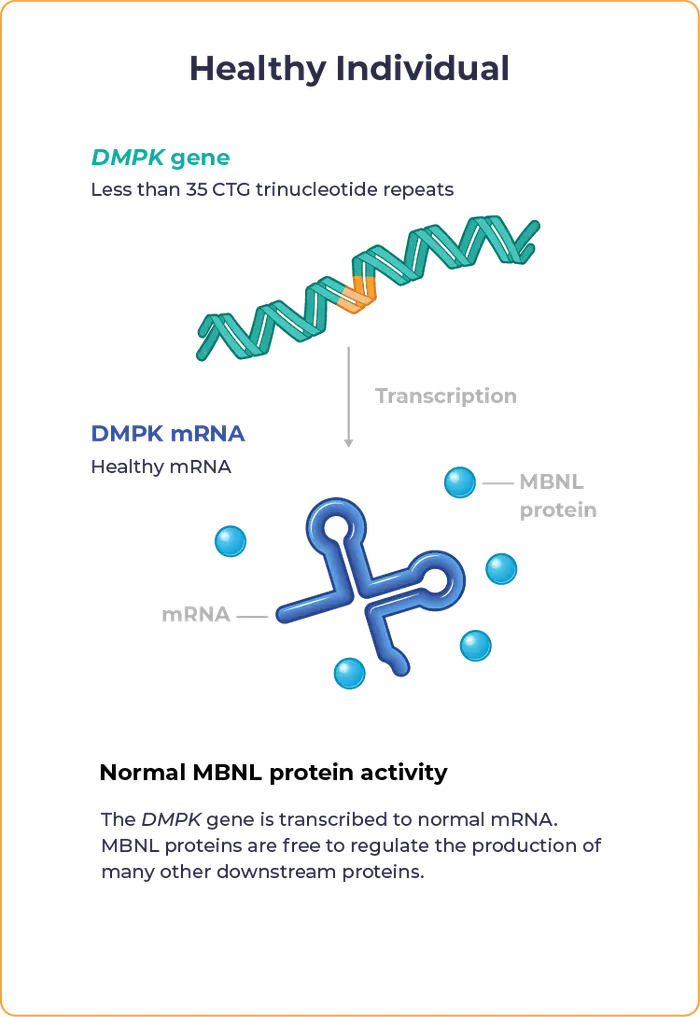
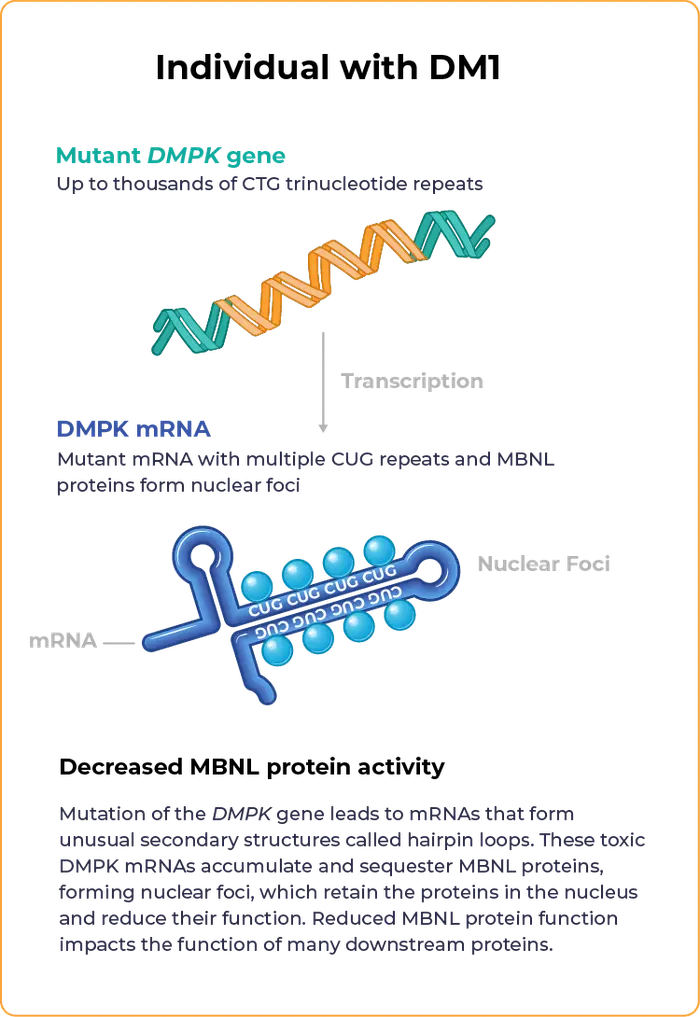
Myotonic dystrophy type 1 – often referred to as DM1 – is a rare, multisystemic disease caused by excessive repeats of the nucleotide sequence CTG within the dystrophia myotonia protein kinase (DMPK) gene. These nucleotide repeats result in toxic mRNA that isolates and reduces function of muscle blind-like (MBNL) proteins, leading to mis-regulation of multiple RNA splicing events that are correlated with DM1 symptoms.
MBNL proteins play an important role in the splicing of mRNAs, essential molecules in the creation of proteins from genetic material.
Decreased MBNL protein activity can lead to slowly progressive muscle wasting, weakness, myotonia and other multi-system defects that worsen over time.
There are currently no approved therapies for DM1. Instead, treatment is largely focused on symptoms management.
Endosomal Escape Vehicle (EEV™)-Oligonucleotides for Myotonic Dystrophy Type 1
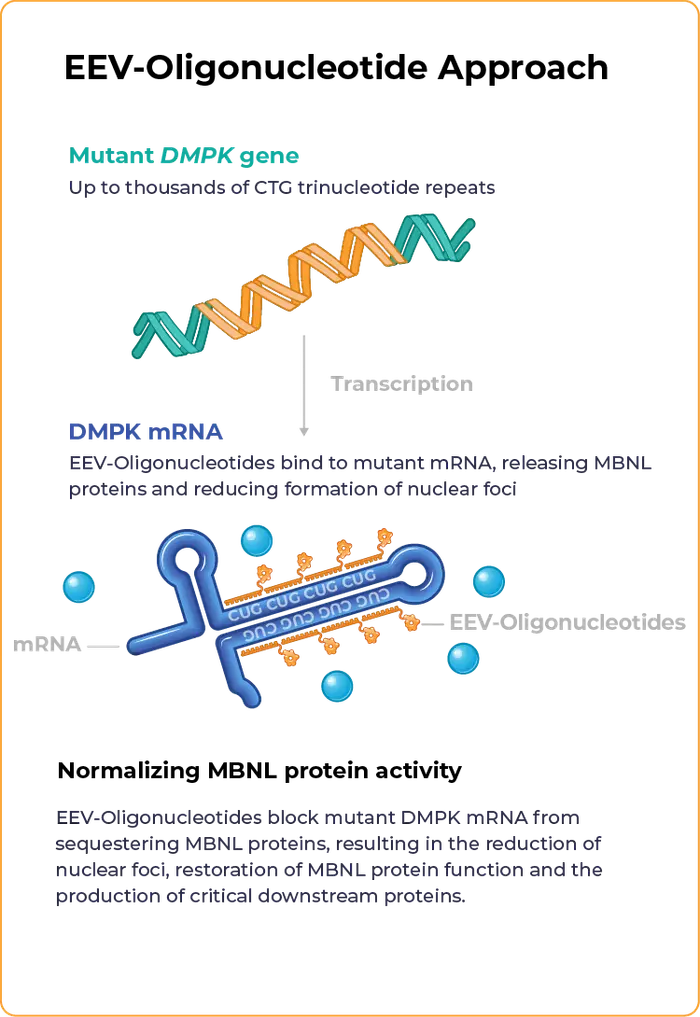
Our proprietary EEV-Oligonucleotide is designed to address the underlying genetic cause of DM1 to restore MBNL protein activity.
In February 2023, Entrada and Vertex initiated a strategic collaboration and license agreement for the development of Endosomal Escape Vehicle-therapeutics for the potential treatment of myotonic dystrophy type 1. The agreement includes a four-year global research collaboration whereby Entrada will continue to advance certain research activities related to VX-670, as well as additional DM1-related research.
Vertex is responsible for global development, manufacturing and commercialization of VX-670 and any additional programs stemming from Entrada’s DM1 research efforts. The program is currently in global Phase 1/2 clinical development, with enrollment and dosing underway. Learn more.
Learn more about our diverse and expanding pipeline or visit our Manuscripts and Presentations pages.
-
1.
Pascual-Gilabert M, López-Castel A, Artero R. Myotonic dystrophy type 1 drug development: a pipeline toward the market. Drug Discovery Today. 2021;26(7):1765-72. doi: 10.1016/j.drudis.2021.03.024.